|
Welcome to Raju's Home Page |
|
|
Completed Projects (I) Formation of Linear Polymers with Pendant
Vinyl Groups via Inclusion Complex Mediated Polymerization of Divinyl Monomers. J. Am. Chem. Soc. 2006, 128,
7752. This project
discusses about the inclusion complexes of β-CDs with divinyl monomers such as ethylene glycol dimethacrylate (EGDMA)
and ethylene glycol methacrylate 4-vinyl benzoate (EGMAVB). The conformational analyses of EGDMA and EGMAVB at various force
fields (MM2*, MM3*, AMBER*, OPLS, MMFF, OPLS2001 and OPLS2003) and density functional theory (B3LYP/6-31G) calculations indicate
that the bent conformation is more stable than the linear one by 3-5 kJ/mol. The interaction energy and stoichiometry of complexes
were estimated by means of docking and molecular dynamics simulations studies. Rigid docking calculations were performed using
Lamarckian Genetic Algorithm (LGA) and molecular dynamics simulations were carried for 2 ns in explicit solvent (TIP3P water
model). The MD simulations of EGMAVB complexation with β-CD revealed that the conformation wherein the styrene end is
outside the cavity is stable. Docking studies and the subsequent binding energies evaluated at the B3LYP/6-31G level of theory
revealed that the complexation of the ligand with the first β-CD has stabilization energies -54.22, -65.93 kJ/mol for
EGDMA and EGMAVB respectively, while addition of second β-CD does not provide substantial stabilization.
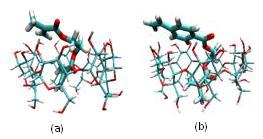
Figure: The lowest energy conformations of (a) EGDMA and (b) EGMAVB obtained through the molecular dynamics simulations.
(II) Hydrogen Bonding in Trivinyl Monomers: Implications for Inclusion Complexation and
Polymerization. Macromolecules 2007, 40, 1824. It discusses about the inclusion complexes of Trimethylolpropane trimethacrylate
(TMPTMA), Trimethylolpropane triacrylate (TMPTA) and Trimethylolpropane diacrylate 4-vinyl benzoate (TMPDAVB) with b-CDs.
The conformational analyses of TMPTA, TMPTMA and TMPDAVB at various force fields (MM2*, MM3*, AMBER*, OPLS, MMFF and OPLS2001)
and density functional theory (B3LYP/6-31G) calculations indicate that the bent conformation of TMPTA is more stable than
linear one by 5-7 kJ/mol, whereas in case of TMPTMA and TMPDAVB stretched conformation
is more stable than bent one by 8-10 kJ/mol. Stoichiometry of trivinyl monomers and b-CD complexes were estimated
by docking protocols, molecular dynamics simulations and density functional theory approaches. Rigid docking calculations
were performed using Lamarckian Genetic Algorithm (LGA) and molecular dynamics simulations were carried for 10 ns in explicit
solvent (TIP3P water model). Docking studies and the subsequent binding energies evaluated at the B3LYP/6-31G level of theory
revealed that TMPTA forms 1:1 complex whereas TMPTMA and TMPDAVB form 1:2 complex with b-CD, corresponding energies
are -93.64, -103.09, and -90.67 kJ/mol respectively. Computational analysis confirmed that in TMPTA hydrogen bonding between
C-H.....O=C brings two acryloyl groups in close vicinity of one another. As a result, both acryloyl groups were included in
the same b-CD
cavity. In case of TMPTMA by the addition of a second b-CD provides substantial stabilization of the order of 46-50 kJ/mol.
In case of TMPDAVB once again formed 1:2 complexes as a result of disruption of hydrogen bonds between two acryloyl groups.
The stoichiometry of inclusion complexes (TMPTA, TMPTMA and TMPDAVB) as well as disposition of the functional groups included
within the b-CD cavity is governed by hydrogen bonding within the monomer.
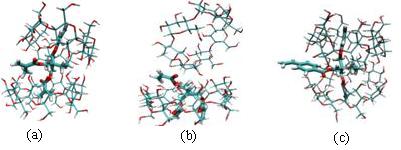
Figure: Conformations obtained from MD simulations (a) TMPTMA (b) TMPTA (c) TMPDAVB.
(III) Active site acidic residues and structural analysis of modelled human aromatase:
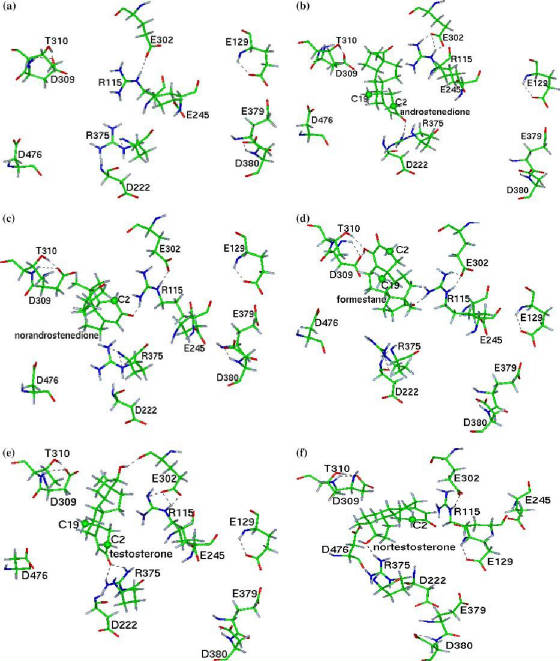
A potential drug target for breast cancer. J. Comp. Aided Mol. Design 2006, 19, 857. This study sheds new light on the role of acidic residues present in the active site cavity of human aromatase. Eight acidic residues (E129, D222, E245, E302, D309, E379, D380 and D476) lining the cavity are identified and studied using comparative modeling, docking, molecular dynamics as well as statistical techniques. The structural environment of these acidic residues is studied to assess the stability of the corresponding carboxylate anions. Results indicate that the environment of the residues E245, E302 and D222 is most suitable for carboxylate ion formation in the uncomplexed form. However, the stability of D309, D222 and D476 anions is seen to increase on complexation to steroidal substrates. In particular, the interaction between D309 and T310, which assists proton transfer, is found to be formed following androgen/nor-androgen complexation. The residue D309 is found to be clamped in the presence of substrate which is not observed in the case of the other residues although they exhibit changes in properties following substrate binding. Information entropic analysis indicates that the residues D309, D222 and D476 have more conformational flexibility compared to E302 and E245 prior to substrate binding. Interaction similar to that between D476 and D309, which is expected to assist androgen aromatization, is proposed between E302 and E245. The inhibition of aromatase activity by 4-hydroxy androstenedione (formestane) is attributed to a critical hydrogen bond formation between the hydroxy moiety and T310/D309 as well as the large distance from D476. The results corroborate well with earlier site directed mutagenesis studies.
Figure: The active site acidic residues when complexed to ligands. (A) is the active site prior to substrate binding.
(IV) Comparative Study on Formamide - Water Complex. Int. J. Quantum Chem. accepted. The hydrogen bonding of 1:1 complexes formed between formamide and water molecule have been investigated
systematically using Hartree-Fock(HF), hybrid density functional theory (B3LYP) and
post-Hartree-Fock (MP2 and CCSD(T)) methods with range of basis sets 6-31G(d) , cc-pVXZ(X = D, T, Q) and aug-cc-pVYZ
(Y = D, T). Three stable structures are considered on the potential energy surface of formamide and water system. The optimized
geometric parameters and interaction energies for various isomers at different levels are estimated. The IR frequencies, intensities
and frequency shifts are reported. The present study shows that B3LYP/aug-cc-pVDZ method gives better performance for formamide-water
complexes.
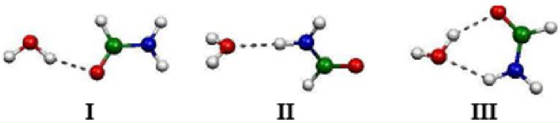
Figure:
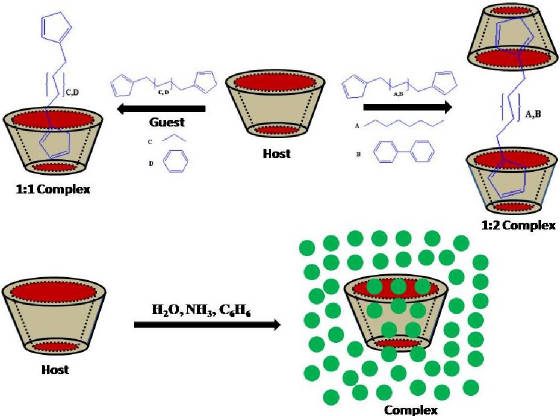
Formamide-water complexes considered in this study. (V) Theoretical studies on inclusion complexes of cyclodextrins. J. Phys. Chem. A
accepted. Quantum chemical calculations and molecular dynamics simulations are carried out to study the host-guest
inclusion complexes of cyclodextrins (a-, b- and g-CD) with small guest molecules such as H2O, NH3,
NH4+, C6H6 and bisimidazolyl compounds. The uptake ability of the CDs to accommodate
the small molecules inside the cavity is examined by sequential addition of 10 molecules of water or ammonia using semiempirical
(PM3) method. In case of benzene, this was done up to 6 molecules. PM3 calculations indicate that a-, b- and g-CD
can accommodate 3, 7, 9 water molecules respectively. In case of NH3 as guest molecule, a-, b-, and g-CDs
can accommodate up to 2, 5, 6 molecules respectively. Semiempirical calculations indicate that two benzene molecules can be
accommodated in a-CD cavity whereas b-,
g-CD cavities adopt 3 and 4 benzene molecules respectively. Molecular dynamics
simulations were carried out for 1.0 ns on benzene and bisimidazolyl complexes of CDs in explicit solvent (TIP3P water model).
The relative binding energies calculated by MM/PBSA method reveal that ligand 1,6-bis(imidazol-l-lyl) hexane (B) and 1,4-bis(imidazol-l-lylmethyl)
benzene (C) molecules prefer to form 1:1 complexes with a-, b- and g-CDs. However C preferentially forms 1:2 complexes with a-CDs. Ligand 1,10-bis(imidazol-l-lyl) decane (A) and 4,4`-(bis(imidazol-l-ylmethylene))biphenyl
(D) form 1:2 complexes with a-, b- and
g-CDs in head-to-head (HH) orientation of CDs. The stability of inclusion compounds
depends on the type of CDs and physicochemical properties of the involved guest. Both these methods (semiempirical and MD
simulations) reveal that b-CDs form more stable complexes as compared to a-, g-CDs with C, D, NH4+
and C6H6 whereas a-CD forms more stable complexes
with A and B.
Figure: Schematic representation of formation of inclusion complexes. (VI) Addition Fragmentation
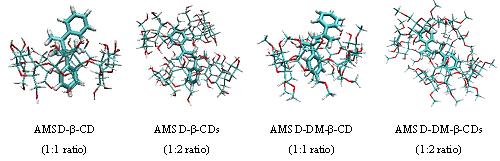
Chain Transfer by AMSD Inclusion Complex: Implications for Macromonomer Synthesis. submitted. This project discusses about the inclusion complexes of a-methyl styrene dimer (AMSD) with b-CD, 2, 6 dimethyl b-CD (DM-b-CD) and the activity of CH=CH2 group of styrene in presence and absence of CDs. The conformational
analysis of AMSD reveals that stretched conformation is more stable than bent one by 2-3 kcal/mol at various levels of theory
(AM1, PM3, HF/6-31G, B3LYP/6-31G and MP2/6-31G). Stability of the complexes is evaluated by using docking studies, molecular
dynamics simulations and density functional theory approaches. Rigid docking calculations were performed using Lamarckian
Genetic Algorithm (LGA) and molecular dynamics simulations were carried for 10 ns in explicit solvent (TIP3P water model).
Docking studies and subsequent binding energies evaluated at B3LYP/6-31G level of theory reveals that the complexation of
the ligand results in substantial stabilization of the order of 3-5 and 2-4 kcal/mol with the first b-CD and DM-b-CD respectively; while the addition
of second b-CD and DM-b-CD
does not provide substantial stabilization. Activity of CH=CH2 is explained from molecular dynamics trajectory
by the disposition of the allyl group within the CD cavity. Quantum chemical descriptors such as natural bond orbital analysis
(NBO), natural population analysis (NPA), and Fukui functions calculated at HF/3-21G level of theory on AM1 optimized geometries,
specify that the activity of R-CH=CH2 group is in the order, AMSD > AMSD-DM-b-CD > AMSD-b-CD.
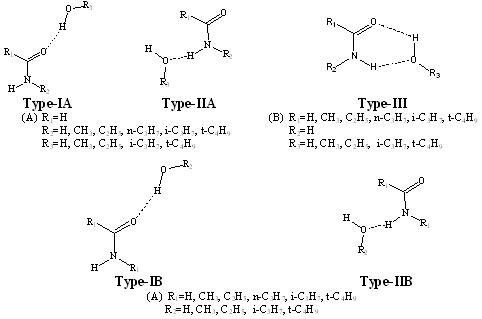
(VII) Effect of alkyl substitution on H-bond strength of substituted formamide-water complexes. Submitted. This project systematically explores the effect of alkyl substitution (CH3, C2H5,
n-C3H7, i-C3H7, and t-C4H9) on the hydrogen bond strengths
(H-bond) of substituted formamide-water complexes. B3LYP/aug-cc-pVDZ method was applied to a total of 215 alkyl substituted
formamide-water complexes to delineate the effect of substitution on the H-bond strength. Complexes are classified into five
types depending on the hydrogen donor, acceptor and the site of alkyl substitution (Type-IA, Type-IIA, Type-IB, Type-IIB and
Type-III). The strength of H-bond was correlated with geometrical parameters such as proton-acceptor (H....Y) distance, the
length of proton donating bond (X-H). In all the complexes N-H and O-H stretching
frequencies are red-shifted. The effect of alkyl substitution on N-H and O-H stretching frequencies were analyzed. Topological
parameters like electron density at H....Y and X-H bond critical points as derived from atom in molecules (AIM) theory was
also evaluated.
Figure: Systems considered in the present study. |